Bosma arini mikroftalmi sendromu
Bosma arini mikroftalmi sendromu (arhinia microphthalmia), otosomal dominant yolla aktarılan kalıtsal bir sendromdur; göz ve burun bulguları ön plandadır.[1][2][3]
Gözler arası açıklık fazladır (hipertelorizm). Belirgin bir mikroftalmi (göz küresi küçüklüğü) ile iris, retina ve göz siniri (n.opticus) defektleri vardır. Katarakt olabilir. Gözyaşı kanalı (nazolakrimal kanal) dardır ya da oluşmamıştır. Kulak malformasyonları ve işitme sorunları saptanır. Burunda malformasyonlar vardır ve burun kemiklerinin çoğu eksiktir. Paranazal sinüslerde hipoplazi görülür. Kafa tabanındaki koku alma merkezi (bulbus olfactorius) gelişmemiştir; bu nedenle, koku alma bozukluğu vardır. Solunum güçlüğü olabilir. Yüz orta bölümünde hipoplazi görülebilir; çukur damak ve/veya damak yarığı bulunur; üst dudak yarığı ender bulgulardan biridir. Dişlerde hipoplazi, hipodonti ve maloklüzyon vardır.[4]
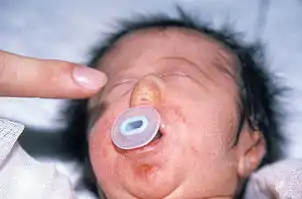
Hipogonadotropik hipogonadizm bulguları saptanır; FSH, LH, testosteron ve östrojen düzeyleri düşüktür. Erkek çocuklarında mikropenis ve skrotuma inmemiş testisler, kız çocuklarında labium majör hipoplazisi ve primer amenore (adet görmeme) olabilir. Kemiklerin mineralizasyonu yetersizdir (osteopeni).[1][2][3][4]
Kaynakça
- Thiele H, Musil A, Nagel F, Majewski F. Familial arhinia, choanal atresia, and microphthalmia. American Journal of Medical Genetics, 63: 310-313, 1996
- Olsen OE, Gjelland K, Reigstad H, Rosendahl K. Congenital absence of the nose: a case report and literature review. Pediatric Radiology, 31: 225-232, 2001
- Graham JM Jr, Lee J. Bosma arhinia microphthalmia syndrome. American Journal of Medical Genetics, 140A: 189-193, 2006
- Brasseur B, Martin CM, Cayci Z, et al. Bosma arhinia microphthalmia syndrome: clinical report and review of the literature. American Journal of Medical Genetics, 170A: 1302-1307, 2016