Cornelia de Lange sendromu
Cornelia de Lange sendromu (CdLS1; Brachmann-de Lange sendromu), 5 fenotipi olan kalıtsal bir sendromdur (1, 3 ve 4 fenotipler otosomal dominant; 2 ve 5 fenotipler X-kromozomu aracılığıyla dominant yolla aktarılır). Tüm fenotiplerde benzer bulgular saptanır; farklılık etkilenen genlerdedir.[1][2][3][4]
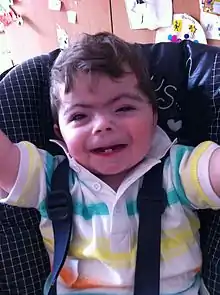
Genel bir gelişme geriliği vardır. Mikrosefali ve brakisefali saptanır. Frontal ve oksipital kemikler düzdür (plagiosefali).[5][6] Ensedeki saç çizgisi aşağıdadır. Boyun kısadır. Kaş çıkıntılarının tümsekleri yüksektir. Hipertrikoz belirgindir; kirpikler ve kaşlar uzun ve kalındır, kaşlar arasında boşluk yoktur. Gözler küçüktür (mikroftalmi). Göz kapakları kabarıktır. Strabismus, nistagmus, ptozis başlıca göz bulgularıdır; optik atrofi nedeniyle görme sorunları vardır. Kulaklarda malformasyonlar, otitis media ve işitme sorunları belirlenir. Burun basıktır, delikleri yukarı dönüktür. Göz, burun ve ağız çevresinde mavimsi renkli pigmentasyon vardır. Filtrum uzun ve belirgindir. Dudaklar ince, kommisuraları aşağı dönüktür. Altçene hipoplaziktir (mikrognati) ve yüzeyinde kemik tümsekleri hissedilir. Dar ve aşırı çukur olan damakta yarık saptanır. Dişler küçüktür (mikrodonti) ve sürme (erüpsiyon) sorunları vardır; bu bulgular kapanış problemlerine (maloklüzyon) ve diastemalara yol açar. Deride damarlı mermer görünümü (cutis marmorata) vardır.[7][8][9]
Omurga sistemi malformasyonuna bağlı skolyoz saptanır. El ve ayaklar küçük, el parmakları kısa ya da eksiktir; sindaktili saptanabilir. Dirsek hareketleri kısıtlıdır.
Konjenital kalp anomalileri, konjenital diyafram hernisi ve batın hernileri, pnömoni riski, üriner sistem anomalileri, meme uçlarında hipoplazi, genital organların hipoplazisi, erkek çocuklarında skrotuma inmemiş testisler ve trombositopeni olası bulgulardır.[10] Kendine yönelik şiddet ve epilepsi bulguları saptanır. Ses incedir, ağlama sesi hırıltılıdır.
Kaynakça
- Jackson L, Kline AD, Barr MA, Koch S. de Lange syndrome: a clinical review of 310 individuals. American Journal of Medical Genetics, 47:940–946, 1993
- Bhuiyan ZA, Klein M, Hammond P, et al. Genotype-phenotype correlations of 39 patients with Cornelia De Lange syndrome: the Dutch experience. Journal of Medical Genetics, 43:568–575, 2006
- Mannini L, Cucco F, Quarantotti V, et al. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Human Mutation, 34(12):1589-1596, 2013
- Yuan B, Pehlivan D, Karaca E, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. Journal of Clinical Investigation, 125(2):636-51, 2015
- Clark DM, Sherer I, Deardorff MA, et al. Identification of a prenatal profile of Cornelia de Lange syndrome (CdLS): a review of 53 CdLS pregnancies. American Journal of Medical Genetics A, 158A:1848–1856, 2012
- Kaiser FJ, Ansari M, Braunholz D, et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Human Molecular Genetics, 23:2888–900, 2014
- Boyle MI, Jespersgaard C, Brøndum-Nielsen K, et al. Cornelia de Lange syndrome. Clinical Genetics, 88(1):1-12, 2015
- Gorlin RJ, Cohen MM Jr, Hennekam RC. Syndromes of the Head and Neck. 4th ed., Oxford University Press, New York, 2001
- Cohen MM Jr. Malformations of the Craniofacial Region: Evolutionary, Embryonic, Genetic, and Clinical Perspectives. American Journal of Medical Genetics (Seminars in Medical Genetics), 115:245-268, 2002
- Schrier SA, Sherer I, Deardorff MA, et al. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. American Journal of Medical Genetics A, 155A: 3007-3024, 2011