Donohue sendromu
Donohue sendromu (diğer adıyla Leprechaunizm; Patterson–Davis sendromu), otosomal resesif yolla aktarılan, nadir bir kalıtsal sendromdur. Leprechaunizm ismi, hastalıktan muzdarip olanların çoğu kez elfvari özelliklere sahip olmaları ve olağandan küçük olmalarından gelir (cüce cin sendromu). Ön planda, insülin reseptör defektine bağlı insülin resistansı bulguları saptanır. Pankreasta adacık beta hücrelerinin hiperplazisi nedeniyle kan insülin düzeyi yüksektir (hiperinsülinemi). Spontan abortus ve bebek ölümü riski oldukça yüksektir.[1][2][3][4]
| Donohue sendromu | |
|---|---|
 | |
|
insülin alıcısı | |
| Uzmanlık |
Endokrinoloji, Romatoloji, Medikal genetik |
Sunuş
Cücelik düzeyinde genel gelişme geriliği vardır. Gözler iridir ve peri yüzü izlenimi alınır. Düşük seviyeli kulak, genişlemiş burun delikleri, büyük ağız açıklığı büyük (makrostomi) ve kalın dudaklar önemli bulgulardır. Dişeti büyümeleri (gingival hiperplazi) belirgindir.[1][3]
Lenf düğümlerinin sayısı azdır. Karaciğerde fibrozis ve safra stazı bulguları vardır.[1][3]
Memelerde hiperplazi, büyük penis ya da klitoris, kistik ovaryumlar saptanır. Aşırı kıllanma (hipertrikoz), deride acanthosis nigricans’lar ve ince bir subkutan yağ dokusu bulguları izlenir. Kas dokusu yeterince gelişemez. Eller ve ayaklar büyüktür.[1][3]
Bozukluğa neden olan mutasyon insülin alıcısını etkilediği için, hasta olanlar insülin insüline dirençlidirler. Buna ek olarak, hipoglisemi ve hiperinsulinemi (kanda çok yüksek seviyede insülin olması) gözlenmektedir. Bozukluk aynı zamanda deri altındaki adipoz dokusunun ciddi bir biçimde azalmasına da neden olur. (Hastaların alışılmadık dış görünümüne bu da neden olmaktadır).[1][3]
Hastalığın bir ölçüde insülin direnci olan ama normal gelişme ve deri altı yağ dağılımının göründüğü daha ılımlı bir biçimi de bilinmektedir.[3] Aynı genin daha az şiddetli bir mutasyona maruz kalmasıyla oluşur.
Kalıtım
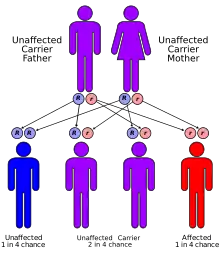
Donohue sendromu otozomal çekinik kalıtsal bir bozukluktur. Bozukluğa neden olan mutasyonlar reseptör moleküllerin üretiminden sorumlu INSR geninin (insülin alıcısı) kodu içindeki pasif kısa kollu 19. (19p13.2) kromozomdadırlar .[2] İnsülin alıcısının işlevselliğini bozan herhangi bir mutasyon benzer etkilere sahip olacağından, hastalıktan sorumlu olabilecek birkaç mutasyon vardır. INSR geni, 1382 amino asitten oluşan bir protein için 22 ekson kodlaması içeren 120000 baz çifti üzerindedir.[5] İntronların bazıları hücrenin çeşidine bağlı olarak uç uca eklenebilir ya da eklenmeyebilir.[6]
Etimoloji
Donohue sendromu ilk olarak 1948'te Kanadalı patolog William L. Donohue (1906-) tarafından tanımlandı.[7] Leprechaunizm ismi, bazı hasta ebeveynleri tarafından küçük düşürücü algılandığı için tamamen terk edildi.[8]
Ayrıca bakınız
- Rabson-Mendenhall sendromu
- Patterson pseudoleprechaunizm sendromu
- Williams sendromu
Kaynakça
- Cantani A, Ziruolo MG, Tacconi ML. A rare polydysmorphic syndrome: leprechaunism--review of forty-nine cases reported in the literature. Annales de Genetique, 30: 221-227, 1987
- Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D (2002). "Genotype-phenotype correlation in inherited severe insulin resistance". Hum. Mol. Genet. 11 (12). ss. 1465-75. doi:10.1093/hmg/11.12.1465. PMID 12023989.
- al-Gazali LI, Khalil M, Devadas K (1993). "A syndrome of insulin resistance resembling leprechaunism in five sibs of consanguineous parents". J. Med. Genet. 30 (6). ss. 470-5. doi:10.1136/jmg.30.6.470. PMC 1016418 $2. PMID 8326490.
- Rosenberg, A. M., Haworth, J. C., Degroot, G. W., Trevenen, C. L., Rechler, M. M. A case of leprechaunism with severe hyperinsulinemia. American Journal of Diseases of Children, 134: 170-175, 1980
- NCBI Sequence Viewer v2.0
- Benecke H, Flier JS, Moller DE (1992). "Alternatively spliced variants of the insulin receptor protein. Expression in normal and diabetic human tissues". J. Clin. Invest. 89 (6). ss. 2066-70. doi:10.1172/JCI115819. PMC 295926 $2. PMID 1602013.
- Donohue WL; Edwards, HE (1948). "Dysendocrinism". J. Pediat. 32 (6). ss. 739-48. doi:10.1016/S0022-3476(48)80231-3. PMID 18866943.
- Şablon:OMIM
| Sınıflandırma |
|---|