Mikroftalmi sendromu
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir.[1][2][3][4] Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.[5]
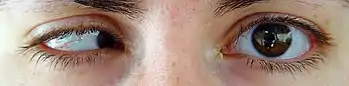
Gözle ilgili (çoğu fenotipte saptanan) bulgular şunlardır: mikroftalmi, anoftalmi ya da displazi (çoğunlukla tek gözde), göz bileşenleriyle ilgili çok sayıda defektler (koloboma), mikrokornea, strabismus, nistagmus, ptozis, katarakt, kaş ve göz kapakları malformasyonları, optik sinir hipoplazisi.[2][3]
İskelet sistemi bulguları arasında kranyum malformasyonları, bazı fenotiplerde cücelik düzeyine yaklaşan gelişme geriliği, toraks, kaburga (kosta), vertebra ve el-ayak parmakları malformasyonları görece sıktır.[3][6]
Maksillofasiyal bölgede, kulak anomalileri, yüz orta bölüm hipoplazisi, küçük çeneler (mikrognati), altçenenin geride olması (mikroretrognati), çukur damak, yarık damak, dil hipoplazisi (mikroglossi), küçük dilde yarık (uvula bifida) olabilir. Dişlerle ilgili bulgular fenotip 2'de (oculofaciocardiodental sendrom) çok sayıda ve oldukça belirgindir.[7]
Santral sinir sistemi (hipotalamus, corpus callosum, beyin korteksi, serebellum hipoplazisi, optik sistem agenezisi, vd) malformasyonlarından kökenli psikomotor gerilik bulguları, hipotoni, paralizi bulguları görülebilir.[2][3][8]
Adrenal bezlerin hipoplazisi, hipopituitarizm, hipotiroidizm gibi endokrin sistem bulgularına ek olarak dış genital organlarda gelişme geriliği, uterus anomalileri, sindirim sistemi anomalileri, böbreklerde hipoplazi farklı fenotiplerde değişen düzeylerde saptanan bulgulardır.[1][3][8][9]
Mikroftalmi ile birlikte maksillofasiyal yarık saptanan başkaca sendromlar arasında Fryns mikroftalmi sendromu ve Bosma arini mikroftalmi sendromu örnekleri verilebilir.
Kaynakça
- Nolen LD, Amor D, Haywood A, et al. Deletion at 14q22-23 indicates a contiguous gene syndrome comprising anophthalmia, pituitary hypoplasia, and ear anomalies. American Journal of Medical Genetics, 140A: 1711-1718, 2006
- Bakrania P, Efthymiou M, Klein JC, et al. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and Hedgehog signaling pathways. American Journal of Human Genetics, 82: 304-319, 2008
- Ng D, Hadley DW, Tifft CJ, Biesecker LG. Genetic heterogeneity of syndromic X-linked recessive microphthalmia-anophthalmia: is Lenz microphthalmia a single disorder? American Journal of Medical Genetics, 110:308–314, 2002
- Esmailpour T, Riazifar H, Liu L, et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. Journal of Medical Genetics, 51: 185-196, 2014
- Okumus, N., Zenciroglu, A., Demirel, N., Bas, A. Y., Ceylaner, S. Apparent Lenz microphthalmia syndrome: a patient with unusual manifestations. Genetic Counselling, 19: 177-182, 2008
- Forrester S, Kovach MJ, Reynolds NM, et al. Manifestations in four males with and an obligate carrier of the Lenz microphthalmia syndrome. American Journal of Medical Genetics, 98(1):92-100, 2001
- Gorlin RJ, Marashi AH, Obwegeser HL. Oculo-facio-cardio-dental (OFCD) syndrome. American Journal of Medical Genetics, 63: 290-292, 1996
- Hilton E, Johnston J, Whalen S, et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. European Journal of Human Genetics, 17:1325–1335, 2009
- Morini F, Pacilli M, Spitz L. Bilateral anophthalmia and esophageal atresia: report of a new patient and review of the literature (Letter). American Journal of Medical Genetics, 132A: 60-62, 2005