Meckel sendromu
Meckel sendromu (Meckel-Gruber sendromu), birden fazla etkisi olan bir genle ilgili (pleiotropic), otosomal resesif yolla aktarılan, 15 kadar fenotipi bulunan kalıtsal bir sendromdur.[1][2] Çok sayıda ve karmaşık bulguların varlığı tanıda güçlükler oluşturur.[3] Bazı uzmanlar, Meckel sendromunun 3 temel bulgusu olduğunu savunurlar: (i) Santral sinir sistemi malformasyonları (özellikle okisipital ensefalosel); (ii) Kistik böbrekler; (iii) Karaciğer patolojileri.[4] Bebeklerin yaşama yetisi çok kısıtlıdır; canlı doğabilenlerin çoğu birkaç hafta içimde yitirilir.[3][4]
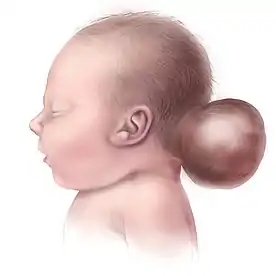
Meckel sendromu (fenotip 1)
Mikrosefali ve holoproensefali saptanır. Oksipital ensefalosel en tipik bulgulardan biridir. Boyun kısa ve kalındır, yelkensi çıkıntılar izlenir (sfenks boynu). Alın bombesi yüksektir. Gözler küçüktür (mikroftalmi), hipertelorizm ya da hipotelorizm (hipertelorizmin zıttı) vardır. İris defekti (koloboma) ile retinal displazi belirlenir. Potter yüzüne özgü bulgular belirgindir. Ağız açıklığı büyüktür (makrostomi), altçene küçüktür (mikrognati). Yenidoğanın sürmüş dişleri vardır (natal dişler). Dudak ve damak yarıkları ile dilde lobülasyon saptanır.[3][4][5][6][7]
Epiglot yarıktır. Akciğerler tam gelişemez (pulmoner hipoplazi). Dolaşım sistemi defektleri arasında atrial ve ventriküler septal defektler, aort koarktasyonu ve patent ductus arteriosus sıkça görülür. Karaciğer bulguları arasında safra yollarında aşırı artma ve genişlemeler ile karaciğer fibrozisi önemlidir. Dalak anomalileri görülür (dalak yokluğu/büyük dalak/aksesuar dalak).[3][4][5][6][7]
İki tane olması gereken göbek kordonu atardamarı (A.umblicalis) tektir. Bağırsakların yerleşimi düzensizdir. Anüs açıklığı oluşmamıştır (imperfore anüs). Dış genital organlar küçük ve biçimsizdir. Erkeklerde testisler skrotuma inmemiştir. Kızlarda vajina ve uterus anomalileri saptanır. Böbrek eksikliği (renal agenezis), polikistik böbrek ve üriner kanal malformasyonları belirgindir, mesane gelişememiştir (hipoplazi).[3][4][5][6][7]
Uzun kemikler eğridir. Parmaklarda sindaktili ve kıvrıklıklar görülür. Adrenal bezlerin gelişimi yetersizdir (hipoplazi).[3][4][5][6][7]
Beyinde oluşum kusurları vardır: Oksipital ensefalosel, Arnold-Chiari malformasyonu, Dandy-Walker malformasyonu, beyin ve beyincik hipoplazisi, corpus callosum yokluğu, görme sistemi (optik traktüs) yokluğu, hidrosefalus, bazı fetüslerde anensefali gibi anomalilerdir.[3][4][5][6][7]
Öteki fenotipler
Öteki fenotiplerde okisipital ensefalosel, kistik böbrekler ve karaciğer patolojileri dışında kalan bulguların bir bölümü yoktur ya da hastaların küçük bir bölümünde görülebilmektedir. Örneğin, ağız bulgularının en önemlisi olan dudak ve damak yarıklarına 8 fenotipte (1, 2, 3, 4, 5, 6, 8, 10) rastlanmaktadır.[3][4][5][6][7] Fenotip 2, Joubert sendromu ile aleldir.[8]
Kaynakça
- Salonen R, Paavola P. Meckel syndrome. Journal of Medical Genetics, 35(6):497-501, 1998
- Tallila J, Salonen R, Kohlschmidt N, et al. Mutation spectrum of Meckel syndrome genes: one group of syndromes or several distinct groups? Human Mutation, 30(8):E813-830, 2009
- Alexiev BA, Lin X, Sun CC, Brenner DS. Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Archives of Pathology & Laboratory Medicine, 130(8):1236-1238, 2006
- Logan CV, Abdel-Hamed Z, Johnson CA. Molecular genetics and pathogenic mechanisms for the severe ciliopathies: insights into neurodevelopment and pathogenesis of neural tube defects. Molecular Neurobiology, 43: 12-26, 2011
- Gazioğlu N, Vural M, Seçkin MS, et al. Meckel-Gruber syndrome. Child's Nervous System, 14(3):142-145, 1998
- Opitz JM, Schultka R, Gobbel L. Meckel on developmental pathology. American Journal of Medical Genetics, 140A: 115-128, 2006
- Barisic I, Boban L, Loane M, et al. Meckel-Gruber Syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. European Journal of Human Genetics, 23(6):746-752, 2015
- Bachmann-Gagescu R, Dempsey JC, Phelps IG, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. Journal of Medical Genetics, 52: 514-522, 2015