Klinefelter sendromu
Klinefelter sendromu ya da 47, XXY sendromu; hücre bölünmesi sırasında, eşeysel kromozom düzensizliklerinden kaynaklanan semptomların hasta kişide görülmesi durumudur.
Hastalığı, 1942'de Dr. Harry Klinefelter ilk olarak tanımlamıştır, doktorun adıyla anılan doğuştan gelen bir hastalıktır.
Nedenleri
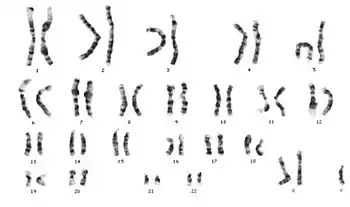
Hücre bölünmesi sırasında eşey kromozomlarından X'in ayrılmaması durumundan kaynaklanan bir sendromdur. İki tane X kromozomu taşıyan bir yumurta hücresinin normal bir sperm ile döllenmesiyle meydana gelir. Normal karyotipte 46, XY olması gereken bireyin, Klinefelter sendromunda 47, XXY şeklinde karyotipi vardır.
Görünümleri
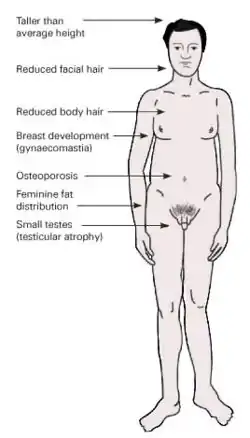
Bu durumdaki kişiler genellikle erkek birey olarak görülür. Puberteyle ortaya çıkar (puberte gecikir). Uzun kol ve bacakları, kadınımsı kalça çıkıntıları ilk olarak göze çarpan özellikleridir. Testisleri küçük, kadınımsı göğüs (jinekomasti) ve kas gelişimleri vardır. Sesleri erkeklere nazaran daha incedir. Hipogonadizm görülebilir. Sakal ve bıyık gelişmleri çok az, vücut kıllanmaları kadınımsı görünümdedir. Spermatogenez görülmez ve kısır bireylerdir.[1]
Canlı erkek doğan bireylerin 500 ya da 1000'inde 1 oranla görülür.
Klinik ve Laboratuvar bulguları[2][3][4]
- Mikrosefali
- Silindirik yüz
- Prognatizm (altçene/üstçene)
- Yarık damak
- Maloklüzyon
- Periodontal patolojiler
- Taurodontism
- Prostat küçük
- Penis normal/hafif küçük
- Varisler
- Ekstragonadal germ hücreli tümör riski (pineal doku, presakral bölge, mesane, prostat, karaciğer)
- Meme kanseri riski
- Psikomotor gelişme geriliği
- Testosteron düzeyi düşük
Türleri
48, XXYY sendromu; yaklaşık 17,000 canlı doğumda bir karşılaşılan Klinefelter sendromunun özelliklerini taşıyan bir hastalıktır. Bu hastalarda semptomlar daha ağır görülür.
Mozaik 47,XXY/46,XY Klinefelter sendromu; şeklinde mozaik olarak da görülebilir. Bu hastalarda semptomlar 47,XXY karyotipli hücrelerin yoğunluğuna bağlı olarak değişir.
Doğum öncesi tanı
Klinefelter sendromu gebelikte tanınabilen bir hastalıktır. Nadir olarak, ultrason bulguları ile fetüs anormal olarak tanımlanabilir. Amniyosentez ve diğer bazı tanı yöntemleri ile Klinefelter sendromununa gebeliklerde kesin tanı konur.
kılavuz
Eylül 2020, Avrupa Androloji Akademisi ilk kez Klinefelter sendromu için kılavuzlar yayınladı.[5]
Ayrıca bakınız
- Turner sendromu
- XYY sendromu
- Triple X sendromu
- 48,XXYY sendromu
Dış bağlantılar
- Ulusal Çocuk Sağlığı Enstitüsü2 Eylül 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- XXYTalk Klinefelter Sendromlular Topluluğu6 Eylül 2008 tarihinde Wayback Machine sitesinde arşivlendi.
- Klinefelter Sendromu ve Dernekleri28 Ekim 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- Amerikan Klinefelter Sendromlular Derneği, Bilgi ve Destek2 Mayıs 2020 tarihinde Wayback Machine sitesinde arşivlendi.
- 47xxy.org4 Ekim 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- Kkinefeltersyndrome.org9 Kasım 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- XXYY Sendromu ve XXYY Projesi7 Ocak 2014 tarihinde Wayback Machine sitesinde arşivlendi.
Kaynakça
- Visootsak J, Aylstock M, Graham JM Jr. Klinefelter syndrome and its variants: an update and review for the primary pediatrician. Clinical Pediatrics (Philadelphia), 40(12):639-651, 2001
- Groth KA, Skakkebæk A, Høst C, et al. Clinical review: Klinefelter syndrome - a clinical update. The Journal of Clinical Endocrinology and Metabolism, 98(1):20-30, 2013
- Haritha A, Jayakumar A. Syndromes as they relate to periodontal disease. Periodontology 2000, 56:65–86, 2011
- Bonomi M, Rochira V, Pasquali D, et al. Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism. Journal of Endocrinological Investigation, 40(2):123-134, 2017
- Zitzmann, Michael; Aksglaede, Lise; Corona, Giovanni; Isidori, Andrea M.; Juul, Anders; T'Sjoen, Guy; Kliesch, Sabine; D'Hauwers, Kathleen; Toppari, Jorma; Słowikowska‐Hilczer, Jolanta; Tüttelmann, Frank (6 Ekim 2020). "European academy of andrology guidelines on Klinefelter Syndrome: Endorsing Organization: European Society of Endocrinology". Andrology (İngilizce): andr.12909. doi:10.1111/andr.12909. ISSN 2047-2919.