Kimyasal kinetik
Reaksiyon kinetiği olarak da bilinen kimyasal kinetik, kimyasal reaksiyonların hızlarını ve mekanizmalarını araştırmakla ilgilenen bir fiziksel kimya dalıdır. Bir sürecin gerçekleştiği yön ile ilgilenen ancak gerçekleşme hızları hakkında bir bilgi vermeyen termodinamik ile karıştırılmamalıdır. Kimyasal kinetik, deneysel koşulların kimyasal reaksiyonların hızı üzerine etkilerini, reaksiyon mekanizmaları ile geçiş hâllerinin verim bilgilerini ve kimyasal reaksiyonların karakteristiklerini tanımlayan matematiksel modellerin çıkarılmasını kapsayan bir bilim alanıdır.
Tarihçe
1864'te Peter Waage ve Cato Guldberg, kimyasal reaksiyon hızının reaksiyona giren maddelerin miktarıyla orantılı olduğunu belirten kütle etki yasasını formüle ederek kimyasal kinetiklerin geliştirilmesine öncülük ettiler.[1][2][3]
Van 't Hoff kimyasal dinamik üzerine çalışmalar gerçekleştirmiş ve 1884'te ünlü "Études de dynamique chimique" (Dinamik Kimya Alanında Çalışmalar) adlı kitabını yayınlamıştır.[4] 1901'de "kimyasal dinamik ve çözeltilerde ozmotik basınç kanunlarının keşfi ile sağladığı müstesna hizmetler karşılığında" ilk Nobel Kimya Ödülü'nü almıştır.[5] Kimyasal kinetik, van 't Hoff'tan sonra hız denklemleri ve hız sabitlerinin türetildiği reaksiyon hızlarının deneysel olarak belirlenmesi ile ilgilenmiştir. Sıfır mertebeli reaksiyonlar (hızın derişime bağlı olarak değişim göstermediği reaksiyonlar) , birinci dereceden reaksiyonlar ve ikinci dereceden reaksiyonlar için görece basit hız denklemleri mevcuttur. Diğer reaksiyonlar için de bu denklemler türetilebilir. Basit (elementer) reaksiyonlar kütle etki yasasına uyar ancak kademeli reaksiyonlar için çeşitli reaksiyon adımlarının hız denklemleri birleştirilmelidir ve bu oldukça karmaşık bir hâl alabilir. Ardışık reaksiyonlarda ise kimyasal kinetiği hızı belirleyen basamak belirler. Birinci dereceden ardışık reaksiyonlar için yatışkın hâl yaklaşımı yapmak hız denklemini basitleştirebilir. Bir reaksiyon için gereken aktivasyon enerjisi Arrhenius ve Eyring denklemi üzerinden deneysel olarak belirlenir. Tepkimeye giren maddelerin fiziksel hâlleri ve derişimleri, reaksiyonda bir katalizör bulunup bulunmadığı ve reaksiyonun gerçekleştiği sıcaklık, reaksiyon hızını etkileyen başlıca unsurlardır.
Gorban ve Yablonsky, kimyasal dinamik tarihinin üç döneme ayrılabileceğini öne sürmüştür.[6] İlki kimyasal reaksiyonların genel yasalarını araştıran ve kinetiği termodinamikle ilişkilendiren van 't Hoff akımıdır. İkincisi de özellikle zincirleme reaksiyonlar için tepkime mekanizmalarının önemine vurgu yapan Semyonov-Hinshelwood akımı olarak düşünülebilir. Üçüncüsü ise Aris'in kimyasal reaksiyon ağlarının ayrıntılı matematiksel açıklamaları ile ilişkilendirilir.
Reaksiyon hızını etkileyen unsurlar
Tepkimeye giren maddelerin doğası
Reaksiyon hızı tepkimeye giren madde (reaktan olarak da adlandırılır) çeşidine göre değişkenlik gösterir. Asit-baz tepkimeleri, tuz oluşumu ve iyon değişimi genelde hızlı gerçekleşen tepkimelerdir. Büyük moleküller oluştuğunda ve moleküller arasında kovalent bağ oluşumu gerçekleştiğinde, tepkimeler genelde yavaşlama eğilimi gösterir.
Tepkimeye giren moleküllerin doğası ve barındırdıkları bağların gücü, reaksiyon hızını önemli ölçüde etkiler.
Fiziksel hâl
Bir reaktanın fiziksel hâli de (katı, sıvı veya gaz) tepkime hızını etkileyen önemli bir unsurdur. Reaktanlar sulu çözeltilerde olduğu gibi aynı fazdayken termal hareket ile birbirlerine yaklaşırlar. Ancak farklı fazlarda bulunuyorlarsa, tepkime reaktanlar arası yüzeyde gerçekleşir. Reaksiyon sadece temas ettikleri bölge ile sınırlı kalır. Örneğin biri sıvı ve biri de gaz hâldeki iki maddenin tepkimeye girmesi durumunda, tepkime yalnızca sıvı yüzeyinde gerçekleşir. Tepkimenin tamamlanması için iyice çalkalamak ve karıştırmak gerekebilir. Bu durum, bir katı veya sıvı reaktan ne kadar iyi ayrışmışsa, birim hacme düşen yüzey alanının ve reaktanların birbiriyle temasının aynı oranda artacağı ve bu sayede reaksiyonun hızlanacağı anlamına gelir. Bir benzerlik kurmak adına şu örnek verilebilir: Ateş yakmak isteyen biri, büyük odun parçaları yerine çok daha ufak dallar ve çıra kullanarak başlayacaktır. Organik kimyada, su yüzeyi reaksiyonları, homojen reaksiyonların heterojen reaksiyonlardan (çözünen ve çözücünün uygun şekilde karışmadığı reaksiyonlar) daha hızlı gerçekleştiği kuralına bir istisnadır.
Yüzey alanı ve katı hâl
Katı bir maddede yalnızca yüzeyde bulunan partiküller reaksiyona girebilir. Bir katıyı daha küçük parçalara ayırmak, yüzeyde daha fazla partikül oluşmasını sağlayarak bu partiküller ile diğer reaktan partiküller arasındaki çarpışma sıklığını arttırmak suretiyle reaksiyonun çok daha hızlı gerçekleşmesine neden olacaktır. Örneğin toz hâldeki şerbet, ince toz hâlinde malik asit (oldukça zayıf bir asit çeşidi) ve sodyum bikarbonat karışımından meydana gelir. Ağızdaki tükürük ile temas ettiğinde, bu kimyasallar hızla çözülür ve reaksiyona girerler. Reaksiyondan ortaya çıkan karbondioksit gazlı içeceklerin verdiği hissi sağlar. Ayrıca havai fişek üreticileri, havai fişeklerdeki yakıtın oksitlenme (yanma) hızını kontrol etmek için katı reaktanların yüzey alanını değiştirir ve bunu çeşitli efektler oluşturmak için kullanırlar. Örneğin, bir havai fişeğin içinde bulunan küçük parçalara bölünmüş alüminyum şiddetli bir şekilde patlar. Daha büyük alüminyum parçaları kullanılırsa, reaksiyon yavaşlar ve yanan metal parçaları çıkarıldıkça kıvılcımlar görülür.
Derişim (konsantrasyon)
Reaksiyonlar tepkimeye giren madde türlerinin çarpışması ile gerçekleşir. Molekül veya iyonların çarpışma sıklığı derişimlerine bağlıdır. Moleküller ne kadar çoksa, birbirlerine çarpıp tepkimeye girme olasılığı da o kadar fazla olur. Dolayısıyla, reaktanların derişimindeki bir artış genellikle reaksiyon hızında artışa neden olurken, derişimdeki bir azalmanın genellikle tam tersine yavaşlatıcı bir etkisi olacaktır. Örneğin yanma tepkimesi saf oksijen bulunan bir ortamda havaya göre (havanın yaklaşık %21'i oksijendir) daha hızlı gerçekleşir.
Hız denklemi, reaksiyon hızının mevcut reaktanların ve diğer türlerin derişimleriyle ilişkisini detaylı olarak gösterir. Matematiksel biçimler reaksiyon mekanizmasına bağlıdır. Belli bir reaksiyon için gerçek hız denklemi deneysel olarak belirlenir ve reaksiyon mekanizması hakkında bilgi verir. Hız denkleminin matematiksel ifadesi şu şekilde gösterilir:

Burada reaksiyon hız sabiti, reaktanının molar derişimi ve ise geçerli reaktanın kısmi reaksiyon sırasıdır. Bir reaktanın kısmi hızı stokiyometrik katsayısı ile genel olarak belirlenemez, sadece deneysel olarak belirlenebilir.




Sıcaklık
Sıcaklığın gerçekleşen tepkime üzerine genellikle büyük bir etkisi vardır. Yüksek sıcaklıkta bulunan moleküllerin termal enerjisi daha fazladır. Her ne kadar yüksek sıcaklıklarda çarpışma sıklığı daha fazla olsa da, bu durum reaksiyon hızındaki artışa esasında oldukça küçük bir katkıda bulunmaktadır. Bu konuda daha önem arz eden durum, reaksiyona girmek için yeterli enerjiye sahip (enerji büyüktür aktivasyon enerjisi: E > Ea) reaktan moleküllerin oranının önemli ölçüde daha yüksek olması ve moleküler enerjilerin Maxwell-Boltzmann dağılımı ile ayrıntılı olarak açıklanabilmesidir.
Sıcaklığın reaksiyon hızına etkisi genellikle formülü ile gösterilen Arrhenius eşitliğine uymaktadır. Burada ön üstel faktör veya A-faktörü, aktivasyon enerjisi, molar gaz sabiti ve mutlak sıcaklıktır.[7]





Belli bir sıcaklıkta kimyasal reaksiyon hızı A-faktörü, aktivasyon enerjisinin büyüklüğü ve reaktanların derişimine bağlıdır. Hızlı gerçekleşen reaksiyonlar genellikle düşük aktivasyon enerjisine sahiptirler.
Hızlı reaksiyonların kinetiği sıcaklık fırlayış yöntemi ile araştırılabilir. Bu metotta sıcaklıkta keskin bir artış kullanılarak dengeye geri dönüş için geçen gevşeme süresi gözlemlenir. Bilhassa kullanışlı sıcaklık fırlayış cihazlarından biri de şok tüpüdür. Şok tüpleri bir gazın sıcaklığını 1000 dereceden yüksek sıcaklıklara hızla çıkarabilir.
Katalizörler
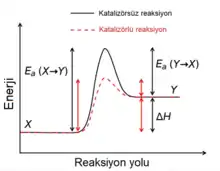
Katalizör bir reaksiyonun hızını değiştiren fakat sonrasında kimyasal olarak değişmeden kalan maddedir. Katalizörler reaksiyon hızını daha düşük bir aktivasyon enerjisine sahip yeni bir reaksiyon mekanizması oluşturarak arttırırlar. Otokatalizde oluşan reaksiyon ürünü, söz konusu reaksiyon için pozitif geri beslemeye yol açan bir katalizör işlevi görür. Biyokimyasal reaksiyonlarda katalizör işlevi gören proteinlere enzim adı verilir. Enzim yoluyla gerçekleşen reaksiyonların hızı Michaelis-Menten kinetiği ile açıklanır. Katalizörler ileri ve geri yöndeki reaksiyonları eşit olarak hızlandırdığından kimyasal dengenin konumunu etkilemezler.
Bazı organik moleküllerde yer değiştiren özel gruplar, komşu grup etkisindeki reaksiyon hızını etkileyebilir.
Basınç
Gaz hâlde gerçekleşen bir reaksiyonda basıncı artırmak reaktanların birbirleriyle çarpışma oranını artırarak reaksiyon hızının artmasına neden olacaktır. Bunun sebebi bir gazın aktivitesinin kısmi basıncıyla doğru orantılı olmasıdır. Bu durum reaksiyon üzerine bir çözeltinin derişiminin artırılmasına benzer bir etki gösterir.
Bu basit kütle etki yasası tesirine ek olarak hız katsayıları da basınç ile değişim gösterebilirler. Gaz karışımına bir inert (tepkimeye girmeyen) gaz eklenirse, hız katsayıları ve yüksek sıcaklıkta gerçekleşen gaz fazdaki birçok reaksiyonun ürünleri değişir. Bu etki üzerindeki değişimlere kimyasal aktivasyon adı verilir. Ekzotermik veya endotermik reaksiyonların ısı aktarımından daha hızlı gerçekleşerek tepkimeye giren moleküllerin termal olmayan enerji dağılımlarına (Boltzmann dağılımına aykırı dağılım) sahip olmasına neden olduğundan dolayı bu olay gerçekleşir. Basıncı arttırmak, reaksiyona giren moleküllerin ve sistemin geri kalanının ısı aktarım hızını arttırarak söz konusu etkinin azalmasını sağlar.
Yoğun fazların da hız katsayıları basınçtan etkilenebilir ancak iyon ve moleküller bu fazda pek sıkıştırılabilir olmadığından ölçülebilir bir etki gözlemleyebilmek için görece yüksek basınçlar uygulamak gerekir. Bu etki sıklıkla hücre tabanlı elmaslar kullanılarak araştırılır.
Bir reaksiyonun kinetiği basınç fırlayış yaklaşımıyla da anlaşılabilir. Bu yöntem ile basınçta hızlı değişiklikler yapıldıktan sonra kimyasal dengeye geri dönüşün gevşeme süresi ölçülür.
Işık emilimi (absorpsiyonu)
Bir kimyasal reaksiyon için gereken aktivasyon enerjisi, tepkimeye giren bir molekülün uygun dalga boyundaki ışığı absorplayarak uyarılmış duruma geçmesiyle sağlanabilir. Işık enerjisi tarafından başlatılan reaksiyonlar fotokimyanın konusudur. Bu reaksiyonların başlıca örneklerinden biri fotosentezdir.
Deneysel yöntemler

Reaksiyon hızlarının deneysel olarak belirlenmesi, reaktan veya ürün konsantrasyonlarının zamanla değişiminin ölçülmesi yoluyla gerçekleştirilir. Örnek olarak, bir reaktanın konsantrasyonu spektrofotometre cihazı ile sistemdeki başka hiçbir reaktan veya ürünün absorbe etmediği bir dalga boyundaki ışık yardımıyla ölçülebilir.
En az birkaç dakika süren reaksiyonlar için reaktanlar istenilen sıcaklıkta karıştırıldıktan sonra gözlem yapmaya başlamak mümkündür.
Hızlı reaksiyonlar
Daha hızlı reaksiyonlar için reaktanların karıştırılması ve belirli bir sıcaklığa getirilmesi için gereken süre, reaksiyonun yarı ömrüne benzer veya daha uzun olabilir.[8] Hızlı reaksiyonları başlatmak için yavaş karıştırma adımı olmayan özel yöntemler şunlardır:
- Durgulu akım yöntemleri karıştırma süresini bir milisaniyeye kadar düşürebilir.[8][9][10] Ancak durgulu akım yöntemlerinin bazı kısıtlamaları vardır. Gazları veya çözeltileri karıştırmak için geçen sürenin durgulu akım yönteminde dikkate alınması gerekir. Yarılanma ömrü saniyenin yüzde birinin altındaysa bu yöntem uygun değildir.
- Önceden karıştırılmış ve başlangıçta dengede olan sistemlerin, sıcaklık fırlayışı ve basınç fırlayışı gibi kimyasal gevşeme yöntemlerinde hızlı ısıtma veya basınçsızlaştırma ile pertürbe edilerek kimyasal dengeden çıkması sağlanır. Dengeden uzaklaşan sistemin tekrar dengeye dönüşünün süresi ölçülür.[8][11][12][13] Örneğin bu yöntem normal koşullar altında 1 μs veya daha düşük bir yarılanma ömrüne sahip (sırasıya hidronyum ve hidroksit iyonları) nötralleşmesini incelemek için kullanılır.[8][13]
- Parlamalı fotolizde bir lazer atımı ile serbest radikaller gibi yüksek derecelerde uyarılmış kimyasal türler üretilir. Sonrasında bu türlerin reaksiyonları üzerinde çalışmalar gerçekleştirilir.[10][14][15][16]

Kimyasal denge
Kimyasal kinetik bir kimyasal reaksiyonun hızı ile ilgilenirken, termodinamik bir reaksiyonun hangi ölçüde gerçekleşeceğini belirler. Tersinir bir reaksiyonda, ileri ve geri tepkime hızları birbirine eşit olduğu (dinamik denge prensibi) ve reaktanlar ile ürünlerin derişimleri değişim göstermediği anda kimyasal dengeye ulaşılır. Bu durum örnek olarak azot ve hidrojenden amonyak üretilen Haber-Bosch sürecinde gözlemlenebilir. Belousov–Zhabotinsky reaksiyonu gibi salınım reaksiyonları, tepkimede kimyasal dengeye ulaşılmadan önce bileşen derişimlerinin uzun bir süre boyu salınım yapabileceğini göstermektedir.
Serbest enerji
Genel olarak incelendiğinde, serbest enerji değişimi (ΔG) kimyasal bir değişimin gerçekleşip gerçekleşmeyeceğini belirlerken, kimyasal kinetik bir reaksiyonun hangi hızda gerçekleşeceğini izah eder. Bir reaksiyon oldukça ekzotermik olsa ve büyük bir entropi değişimi gösterse dahi eğer çok yavaşsa pratikte gerçekleşmez. Bir reaktandan iki ürün elde edilirken eğer tepkime kinetik reaksiyon kontrolü gibi özel koşullarda tutulmuyorsa bu iki üründen termodinamik olarak en kararlı ürün oluşur. Her biri farklı bir ürün oluşturan, karşılıklı olarak hızla dönüşen iki reaktan için ürün oranının belirlenmesinde ise Curtin–Hammett prensibi uygulanır. Serbest enerji ilişkilerinden yola çıkılarak bir tepkimenin reaksiyon hız sabitleri hakkında tahminlerde bulunmak mümkündür.
Reaktanlardan birine ait bir atomun, izotopu ile yer değiştirildiği zaman reaksiyon hızında oluşan farka kinetik izotop etkisi adı verilir.
Kimyasal kinetik, kimya mühendisliğinde reaktörlerde kalma süresi ve ısı transferi hesabı, polimer kimyasında da molar kütle dağılımı hakkında bilgi sağlar. Korozyon mühendisliğine de çeşitli bilgiler sağlamaktadır.
Modeller ve uygulamalar
Reaksiyon kinetiğini tanımlayan matematiksel modeller, kimyagerlere ve kimya mühendislerine gıda bozulması, biyolojik sistemlerin kimyası, mikroorganizmaların üremesi ve stratosferik ozon bozulması gibi kimyasal süreçleri daha iyi anlamak ve tanımlamak için araçlar sağlamaktadır. Bu modeller aynı zamanda kimyasal reaktörlerin ürün verimini optimize etmek, ürünlerini daha verimli bir şekilde ayırmak ve çevreye zararlı yan ürünlerini ortadan kaldırmak için gerekli tasarım ve değiştirmelerde kullanılabilir. Örneğin, kinetik modeller ağır hidrokarbonların benzine ve gaza katalitik olarak parçalanması işleminde, ağır hidrokarbonlardan benzinin en yüksek verimle üretileceği sıcaklığı ve basıncı bulmak için kullanılabilir.
Kimyasal kinetik, eğri uydurma yönteminin ve adi diferansiyel denklem çözümlerinin (ODE çözümü) bir fonksiyonu halindeki özel paketleri modelleme yoluyla sık sık araştırılır ve doğrulanır.[17]
Ayrıca bakınız
- Alev hızı
- Elektrokimyasal kinetik
- Heterojen kataliz
- İçsel düşük boyutlu manifold
- Kimyasal hız sabitlerini deneysel verilere uydurmak için kullanılan Matlab’in PottersWheel araç çubuğu
- Kimyasal reaksiyon mühendisliği
- Korozyon mühendisliği
- MLAB kimyasal kinetik modelleme paketi
- Otokatalitik reaksiyonlar ve düzen oluşturma
- Patlama
- Reaksiyon ilerleyişinin kinetik analizi
- Termal olmayan yüzey reaksiyonu
Dış bağlantılar
- YouTube üzerinde LearnChemE tarafından yayınlanmış kimyasal reaksiyon mühendisliği dersleri
- Kimya uygulamaları4 Haziran 2009 tarihinde Wayback Machine sitesinde arşivlendi.
- Waterloo Üniversitesi
- Gaz fazda gerçekleşen reaksiyonların kimyasal kinetiği
- Kinpy: Kinetik denklemleri çözmek için Python kodu türeticisi
- Reaksiyon hız kanunu ve reaksiyon profili – bir sıcaklık, derişim, çözelti ve katalizör sorusu – bir reaksiyon ne kadar hızlı ilerler (SciFox’tan TIB AV – Portal’daki bir video)
Kaynakça
- C.M. Guldberg and P. Waage,"Studies Concerning Affinity" Forhandlinger i Videnskabs-Selskabet i Christiania (1864), 35
- P. Waage, "Experiments for Determining the Affinity Law" ,Forhandlinger i Videnskabs-Selskabet i Christiania, (1864) 92.
- C.M. Guldberg, "Concerning the Laws of Chemical Affinity", Forhandlinger i Videnskabs-Selskabet i Christiania (1864) 111
- Hoff, J. H. van't (Jacobus Henricus van't); Cohen, Ernst; Ewan, Thomas (1896-01-01). Studies in chemical dynamics. Amsterdam : F. Muller ; London : Williams & Norgate.
- The Nobel Prize in Chemistry 1901, Nobel Prizes and Laureates, official website.
- A.N. Gorban, G.S. Yablonsky Three Waves of Chemical Dynamics, Mathematical Modelling of Natural Phenomena 10(5) (2015), p. 1–5.
- Laidler, K. J. Chemical Kinetics (3rd ed., Harper and Row 1987) p.42 ISBN 0-06-043862-2
- Laidler, K. J. Chemical Kinetics (3rd ed., Harper and Row 1987) p.33-39 ISBN 0-06-043862-2
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.254-256 ISBN 0-07-288362-6
- Atkins P. and de Paula J., Physical Chemistry (8th ed., W.H. Freeman 2006) p.793 ISBN 0-7167-8759-8
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.256-8 ISBN 0-07-288362-6
- Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1999) p.140-3 ISBN 0-13-737123-3
- Atkins P. and de Paula J., Physical Chemistry (8th ed., W.H. Freeman 2006) pp.805-7 ISBN 0-7167-8759-8
- Laidler, K.J. Chemical Kinetics (3rd ed., Harper and Row 1987) p.359-360 ISBN 0-06-043862-2
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.264-6 ISBN 0-07-288362-6
- Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1999) p.94-97 ISBN 0-13-737123-3
- "Chemical Kinetics: Simple Binding: F + G ⇋ B" (PDF). Civilized Software, Inc. Retrieved 2015-09-01.
Kimya mühendisliği konuları | ||
|---|---|---|
| Tarih |  | |
| Kavramlar | ||
| Ünite operasyonları | ||
| Ünite prosesleri | ||
| Dallar |
| |
| Diğer |
| |
Kimyanın dalları | |
|---|---|
Kimya literatürü • Kimyasal formül dizini • Biyomoleküller listesi • İnorganik bileşikler listesi • Periyodik tablo | |
| Fiziksel |
|
| Biyolojik |
|
| Organik |
|
| İnorganik |
|
| Analitik |
|
| Diğer |
|
| Ayrıca bakınız |
|